References
Explore our experiences
We have been at the leading edge of lab automation for 20 years and have developed revolutionary systems for a wide range of application areas. In addition to this, we have optimized products with technical advancements, successfully prepared products for industrialization and serial production, and carried out life cycle management.
Our applications expertise covers a wide range of technologies such as capillary electrophoresis, real-time PCR, Pyrosequencing, and next-generation sequencing. Some of the products we have played a role in are shown below.

 Spatial Transcriptomics
Spatial Transcriptomics
Bridging Molecules and Tissues

Spatial transcriptomics is an innovative technology that enables the analysis of gene expression directly within its spatial context in tissues. It links molecular information to tissue structure and provides a deeper understanding of biological processes. This method is particularly valuable for deciphering the complexity of cell types and their interactions within different tissue environments.
Applications and Significance
This technology is used in cancer research, the analysis of disease mechanisms, and the development of personalized therapies.
It enables the identification of disease-specific patterns and the discovery of new therapeutic targets.
Spatial transcriptomics is revolutionizing the life sciences by bridging the gap between genomics and histology.
 Magnetic Bead Extraction
Magnetic Bead Extraction
Fast and easy purification
A commonly used approach for nucleic acid extraction is based on magnetic bead technology. The sample containing the nucleic acids is mixed with a solid phase consisting of silica-coated magnetic beads. Since the nucleic acids will bind to the beads, they can easily be separated from the remaining solution thanks to their magnetic properties. After the separation, the beads are first washed before the nucleic acids are released from the beads by immersing them into elution solution.
A wide range of starting materials
Magnetic bead technology allows extraction and purification of eukaryotic, bacterial as well as viral DNA and RNA from a wide range of biological materials such as blood, tissues, as well as other type of materials like bacteria, viruses and plants. The purified nucleic acids can be used in downstream applications in biomedical research, molecular diagnostics and gene expression analyses.
For information about a product based on this technology, see EZ1 Advanced XL and QIAsymphony SP.
 Spin-Column Extraction
Spin-Column Extraction
Fast and efficient purification
In molecular biology workflows, spin column applications efficiently extract nucleic acids by first lysing samples to release DNA or RNA.
These nucleic acids are then bound to a silica membrane inside the spin column using a high-salt buffer. Through centrifugation, impurities like proteins are washed away, and the purified nucleic acids are subsequently eluted in a low-salt buffer. This process, using centrifugal force, ensures a clean separation of nucleic acids, ready for subsequent molecular analysis.
 Single Cell Isolation
Single Cell Isolation
Individual and viable cells
Single cell isolation is a prerequisite for a number of medical and biochemical applications, such as RT-qPCR, genotyping, cellular phenotype determination, NGS (RNA sequencing and whole genome sequencing), protein analysis with labeled antibodies and genotyping. For these applications, it is crucial that truly individual cells have been isolated and that these cells are viable. In large scale-studies, it’s also essential to avoid labor-intensive methods. Therefore, various methods and devices, that increase the efficiency of single cell isolation have been developed.
Magnetic technology
One time-saving approach is based on magnetic technology. Here, the cells are cultivated on arrays packed with thousands of magnetic microrafts, where each raft is designed to hold single cells. The cells of interest are identified using a microscope, and the corresponding microrafts are dislodged from the array using a magnetic wand and finally transferred to a test tube or a cell culture dish. This method ensures minimal manipulation of the cells and cell viability for several days, thereby providing a good option for a wide range of downstream applications.
For information about products based on this technology, see QIAscout.
View our reference projects
 Optical Analysis
Optical Analysis
Measuring quantity and quality
Accurate measurement and reliable quality control of DNA, RNA and protein samples is a prerequisite for a multitude of assays in medical research and biochemical applications. By using a spectrophotometer to read the UV/VIS absorption spectrum of a compound in a given sample, it is possible to measure the signal and calculate its quantity. Spectral profiling, i.e. reading the absorption of a sample at a range of different wavelengths, can reveal further information about the quality and the composition of the sample, e.g. detecting impurities. The profiles of different sample types are unique; e.g. a pure RNA sample will give rise to a profile that is decidedly different from an RNA sample spiked with gDNA.
Reproducibility and linearity
In order to ensure that the measurements are reliable, serial dilutions of the samples should be made to calculate the linearity. The regression coefficient should optimally be >0.999, but this is difficult to achieve with manual measurements. Therefore it’s useful to employ automated spectrophotometry systems, providing the high system linearity and reproducibility required for many molecular biology applications.
For information about products based on this technology, see QIAxpert.
Small-scale sample disruption
Analysis of nucleic acids and proteins from biological samples are often a crucial part of medical and biochemical applications. Therefore, a fast and efficient method is needed for releasing these biomolecules from biological samples. Usually mechanical, physical, chemical and/or enzymatic methods are used to disrupt the biological matrixes and cells. In low-throughput workflows, rotor-stator disruption is a suitable mechanical method. It’s carried out using a rotor-stator homogenizer, essentially a handheld high-speed blender, which mechanically shears the samples. The shearing is a result of both: the rotation of the blade and the turbulence in the sample. This method is often combined with enzymatic lysis and/or physical disruption using liquid nitrogen for more efficient disruption and homogenization.
 Capillary Electrophoresis
Capillary Electrophoresis
High resolution separation of biomolecules
Capillary electrophoresis is a method that utilizes narrow capillaries (typically made of silica) under high voltage for the separation of ions, molecules or molecular complexes. In many applications, it can replace traditional slab-gel electrophoresis of nucleic acids and proteins. It can be performed with or without a gel, or with a special polymer solution inside the capillaries, either under native or denaturing conditions. Modern instruments allow full control of all electrophoretic parameters and thus provide high reproducibility, which often is associated with high resolution; thus, in DNA analyses it is often possible to achieve 1 bp resolution. The diameter of the capillaries is generally between 20 and 100 μm and the length 17–100 cm, so that the capillary volume typically is 10–100 μl. The inner wall of the capillaries may be coated to control both the interaction of the analytes with the silica surface and the electroendoosmotic flow. The outer capillary surface is polymer-coated to improve its mechanical stability.
Samples can be loaded into the capillaries either by electrokinetic injection (applying a high voltage for a short, defined period of time) or by hydrostatic pressure. Another difference, in comparison to gel electrophoresis, is that in capillary electrophoresis a detector remains fixed, so that all of the analytes must travel the same distance. The migration time is then used to identify each analyte in every sample. In addition, many samples may be run simultaneously, each sample in its own capillary. The results are usually displayed as electropherograms. The analytes can be detected using a number of detection methods, including UV absorbance, fluorescence, refractive index, etc. Using appropriate standards, the peak area in each electropherogram can be correlated with the amount of each analyte, and thereby its concentration can be determined.
Capillary electrophoresis has many advantages compared to slab gel electrophoresis and HPLC; it gives high efficiency separations, it’s automated, affordable and environment-friendly. In addition, it only requires small sample volumes, it’s fast and easy to perform. Since the sensitivity that can be achieved by capillary electrophoresis is extremely high, the method also allows analysis of samples containing very low analyte concentrations. Different modes of CE, as for example capillary zone electrophoresis, capillary gel electrophoresis, capillary isoelectric focusing and affinity CE, microfluidic CE, provide a large choice of operating modes from which one can select the best suited for a particular analytical application.
Downstream applications
Being a versatile, fast and accurate separation method, capillary electrophoresis can ideally be used for protein and nucleic acid analysis in a number of different applications, involving sequencing, size analyses and quality control. Apart from using it for direct analysis of PCR/RT-PCR amplicons, restriction fragments, cDNA and NGS libraries, it can also be advantageous to include it in workflows involving genotyping, CRISPR, gene expression and regulation, and next-generation sequencing.
For information about a product based on this technology, see QIAxcel Advanced.
 Liquid Handling
Liquid Handling
Automation for accuracy
The increasing demand for reliable high-throughput protocols in medical research and life science applications has created a growing market of automated liquid handling systems. They provide the required accuracy and reproducibility for state-of-the-art PCR, NGS technologies as well as sample preparation for the other downstream processes. Moreover, the automated systems are enabling large-scale studies and saving a lot of time for the laboratory personnel.
Reproducible pipetting
Automated pipetting systems are robotic instruments used to dispense predefined volumes of reagents and samples into vessels such as test tubes or 96-well plates in order to avoid manual intervention thereby ensuring consistent accuracy and reproducibility. These instruments feature robotic hands which handle single or multi-channel pipettes matching tip arrays. Instruments that utilize liquid displacement pipetting technology offer the choice between fixed and disposable tips and a wide range of pipetting volumes. Air displacement technology on the other hand eliminates the need for daily maintenance, since system liquids aren’t required. Today, there are automated pipetting systems offering both of these technologies. The advanced designs of the automated pipetting systems offer safe and reliable pipetting while preventing contamination and carry-over simultaneously that is a prerequisite for most downstream molecular biology applications today.
For information about products based on this technology, see QIAgility, QIAsymphony AS
 Automated Sealing
Automated Sealing
Bead sealing for reproducible qPCR
Maintaining the integrity of qPCR samples is essential, but sometimes difficult to achieve with the designs of qPCR vessels and plate sealing available on the market today. The use of individual tube caps, cap strips or tapes renders a high risk of incomplete sealing of the sample tubes, likely leading to evaporation and contamination of the samples. Hence, a technology which helps eliminating these risks is of great interest. To accommodate this issue, an automated module for dispensing beads into qPCR sample tubes and 96-well plates has been developed. It holds a proprietary bead dispenser (adaptable for a variety of bead diameters), which dispenses beads into each chosen vessel and seals them by a controlled force via a plunger that presses the beads into a vessel, thereby securing maximal tightness. The bead dispenser holds up to 360 pieces of 4 mm diameter beads, which are dispensed at a 100 % dispensation rate, and thus contribute substantially to the high reliability of this module.
Safety and flexibility
The bead sealing device has been designed for maintaining an absolute integrity of the samples. To prevent any degradation of the samples a cooling plate can be positioned below the sample plate. The vessels are efficiently sealed by the beads and an integrated optical sensor can perform a load-check of the beads if needed. The safety of the qPCR system is further ensured with the actual design of the bead sealing module that inherently prevents any cross-contamination. Thereby, the Bead Sealer provides an attractive combination of safety features while simultaneously ensuring a minimum of hands-on tasks. The only manual intervention required is supplying the samples and selecting a suitable program to define the vessels or plates that need to be sealed. It is compatible with different interfaces so that the data can be transferred via cloud, WLAN or Bluetooth. Due to its flexibility and safety features, the bead dispenser can therefore easily be integrated in any existing workflow. The bead sealing module hence provides a good option for improving the efficiency and reproducibility of high-throughput qPCR workflows.
For information about products based on this technology, see TubeSealer.
 Real-Time PCR
Real-Time PCR
Monitoring DNA amplification
Real-time polymerase chain reaction (RT-PCR), also known as quantitative PCR, provides an elegant solution for quantification of DNA sequences in real time, as opposed to conventional PCR, where only the final result of the PCR amplification can be analyzed. The amplification reaction progresses due to repeated cycles of heating and cooling at predefined temperatures for every stage, which are referred to as denaturation, annealing and elongation. The PCR also requires specific reagents, namely DNA template, DNA polymerase, primers (oligonucleotides) serving as starting points and thereby guiding the amplification, as well as dNTPs, serving as building blocks. In order to follow the amplification process in real-time, fluorescent dyes are linked to the primers so that the DNA amplification can be traced; i.e. the fluorescent signals are effectively reporting snapshots of the reaction to the software. The reaction can either be traced with one dye only, e.g. SYBR® Green, or with up to 6 dyes for multiplex RT-PCRs where several DNA targets are amplified simultaneously. This requires a RT-PCR cycler with 6 optical channels, as in the Rotor-Gene Q. Throughout the amplification process, the RT-PCR cycler will send optical data to its software, which will display the results as a curve indicating the amount of signal, i.e. the amount of amplification, in each cycle.
Sensitivity and accuracy
Successful RT-PCR runs rely on high-precision technology; they require a reliable RT-PCR cycler with minimal tube-to-tube variation, fast ramp rate, and optical and thermal accuracy. Different solutions can be used to reach a precise and uniform temperature across all samples in a short period of time during the reaction; for example, Peltier elements and air flow or liquid flow are efficient in controlling and evening out the temperature. The excitation of the fluorescent dyes can be achieved by light sources, such as a halogen, UV, LED lights or a Xenon lamp in combination with dye-specific filters. The signals are captured by a detector (e.g. a photodiode or a CCD), amplified by the photomultiplier and sent to the software.
Downstream applications
Over the years, the technology mentioned above has enabled RT-PCR cyclers to become increasingly sophisticated and reliable, and hence today RT-PCR plays an essential role in many biomedical applications such as gene expression analysis, pathogen detection, DNA methylation analysis, SNP genotyping and gene scanning, as well as miRNA research.
For information about products based on this technology, see Rotor-Gene Q.
 Pyrosequencing
Pyrosequencing
Shedding light on DNA sequences
Pyrosequencing is a high-throughput sequencing-by-synthesis method used to quantify sequence variation. It’s based on a chemical reaction that generates a sequence of light signals, which reflects the sequence of the nascent DNA.
Efficient chemistry
The pyrosequencing reaction relies on the action of four enzymes: DNA polymerase, ATP sulfurylase, luciferase and apyrase. DNA polymerase is used to elongate the DNA, upon which pyrophosphate is released. ATP sulfurylase converts the pyrophosphate to ATP, which is used by the luciferase to oxidize luciferin to oxyluciferin. This reaction generates a light signal, which is detected by a camera and displayed as peaks in a so called pyrogram. At the end of each reaction cycle, apyrase removes any unincorporated nucleotides left in the reaction.
The amount of light released in each pyrosequencing reaction, i.e. the height of each peak, is directly proportional to the number of incorporated nucleotides. If for example 3 nucleotides are incorporated, three ATPs are generated giving rise to triple peak in the pyrogram. The DNA sequence can be determined since the four nucleotides A, C, G and T are dispensed in a predefined order, so that each signal peak can be correlated with a specific nucleotide. This sequencing technology provides quantitative real-time data that can be used for characterization of single nucleotide polymorphisms (SNPs) and insertions-deletions (indels), as well as quantification of allele frequencies and DNA methylation levels.
For information about products based on this technology, see PyroMark.
 Next-generation Sequencing
Next-generation Sequencing
From single DNA sequences to massively parallel sequencing
After over 20 years of conventional Sanger sequencing, next-generation sequencing (NGS) emerged in 2004. With this high-throughput technology, the efficiency of sequencing increased with a significant leap. Rather than merely sequencing single DNA sequences, it was now possible to perform parallel sequencing of many short DNA fragments so that e.g. the whole human genome could be sequenced in less than a week at a fraction of the cost.
Sequencing technologies
Under the NGS label, we can include the so called second, third and fourth generation sequencers. Nowadays, there are several technologies utilizing a variety of methods, as for example:
- Illumina — sequencing by synthesis (bridge amplification) and detection of fluorescence signals
- Ion Torrent — sequencing by synthesis (emulsion amplification) and detection of changes of pH by an ion semiconductor
- Pacific Biosciences — single-molecule real-time sequencer
- 454 Life Sciences — sequencing by synthesis (emulsion amplification) and detection of pyrosequencing signals
- SOLiD — sequencing by ligation (emulsion amplification) and detection of fluorescence signals
- Qiagen GeneReader — sequencing by synthesis (emulsion amplification) and detection of fluorescence signals
- Oxford Nanopore — Nanopore DNA sequencing
Sequencing workflow
NGS workflow is a complex process which involves several steps. A typical workflow includes:
- Nucleic acid extraction
- Fragmentation and quality control (QC)
- Target enrichment (optional) and QC
- Library preparation and QC
- Clonal amplification (optional)
- Sequencing
- Data analysis and interpretation
NGS is used for different applications, such as whole genome and whole exome sequencing, transcriptome profiling, targeted DNA re-sequencing, epigenomics, etc.
NGS workflow key points
Nucleic acid extraction
The origin of the biological material to be studied influences both the amount and the quality of the extracted nucleic acids; i.e. the DNA or RNA can be degraded/compromised to varying degrees. Also, the heterogeneity of the starting material dictates the most suitable workflow to be used for the purpose of the study (e.g. whole genome versus target re-sequencing, sequencing technology and secondary analysis algorithms).
Target enrichment
Depending on the mutations or variants to be investigated, the target enrichment can either be hybridization-based or multiplex PCR-based or simply not implemented at all if whole genome sequencing is required. A target enrichment step normally increases the NGS turnaround time, but on the other hand it allows the analysis to focus more on the genetic traits that really matter. In this way, it is possible to optimize the output of the subsequent NGS platform by allowing higher multiplexing.
Sequencing
In the next step, target libraries or single nucleic acids are subjected to sequencing. For the most common second generation NGS sequencers, the templates are subsequently subjected to reagents containing DNA polymerase and labeled or native dNTPs. Labeled nucleotides generally contain a fluorescent dye, which is different for each base (A, C, G and T). The DNA polymerase incorporates the dNTPs to the ends of the growing DNA strands, which will give rise to patterns of fluorescent signals or released protons reflecting the composition of each DNA sequence of the libraries. The released protons or fluorescent signals are subsequently detected by sensors or high-resolution digital cameras, respectively. The signals are then registered and analyzed by the corresponding NGS sequencer software and the DNA sequences are finally digitalized and compiled in the form of *.fastq files suitable for downstream bioinformatics software analyses.
Library preparation
Library preparation is a multistep procedure, which is specific for each platform. The aim of this step is to ligate sequencer-specific adapters and to distinguish the samples from one another using barcodes; thereby, the sequencer can process more than one sample at a time. Fourth generation sequencers don’t require library preparation, as the templates don’t need to be mobilized on a solid substrate, but on the other hand they don’t allow multiplexing of samples in a single run.
Clonal amplification
For second generation NGS technologies (e.g. the Qiagen GeneReader platform), the last step before sequencing is a so called clonal amplification. Sequencing libraries are immobilized on a solid substrate (flow cell or beads) and clonally amplified to allow signal detection during sequencing. Clonally amplified targets are then hybridized with sequencing primers to allow base incorporation during the sequencing step. This step isn’t required for third (e.g. PacBio) and fourth (Oxford Nanopore) generation NGS technologies, which perform single nucleic acid sequencing.
Data analysis and interpretation
The final results (i.e. *.fastq files) are further processed by dedicated bioinformatic pipelines, which present the change in the genomic sequence of each biological sample and compares it to a given reference sequence. Alternatively, the data can be used to build up a new reference sequence. When studying genomic mutations, the observed variations may eventually be associated with pathogenic variations, such as somatic variations detected in cancer cells, germline mutations associated with inherited diseases and other genetically associated diseases.
For information about an NGS workflow based on this technology, see Qiagen GeneReader NGS.
 Flow Cytometry
Flow Cytometry
Enhancing Cell Analysis with Flow Cytometry
Flow cytometry is a powerful laboratory technique for analyzing and counting individual cells and particles in a liquid suspension. There are different techniques such as laser-based or impedance-based flow cytometry. In impedance-based flow cytometry, a sample is passed through a flow cytometer in which the cells pass through an electromagnetic field (impedance) in very small channels in order to analyze their properties. This method is widely used in biology, immunology and clinical diagnostics for tasks such as cell sorting and characterization of cell populations.
 eviFluor Duo Fluorometer
eviFluor Duo Fluorometer
Product Highlights
- High-sensitivity, dual-channel fluorescence detection integrated seamlessly into the liquid handler workflow
- High accuracy and reliability thanks to consistent automated workflow and precise micro-cuvette technology
- Up to 90% reduction in reagent consumption
- Increased data integrity with automatic sample identification, data capture and transfer, ready for downstream processing
- Space-saving design with compact SBS footprint
The Task
High-quality samples are crucial for successful nucleic acid analysis in PCR and next-generation sequencing (NGS) applications. Our task was to develop a module that meets the growing need for seamless integration of quantification and quality measurements into liquid handlers.
To increase workflow efficiency and reduce process costs, the device needed to use minimal volumes of reagents and sample material, while maintaining the accuracy and precision of conventional instruments.
The Challenge
Current methodologies for sample-quality measurements require relatively large volumes of dye reagents, which significantly impacts overall costs. One of the key challenges we faced was minimizing the volume of both sample material and reagents needed for quantification. In addition, we developed advanced technology and algorithms to achieve high precision and accuracy in the measurements.
Since manual quality measurements are time-consuming, it was also essential to increase workflow efficiency and speed by integrating the measurement into the liquid handler workflow.
The Solution
The eviFluor Duo Fluorometer is a compact, dual channel fluorometer that enables in-process DNA and RNA quantification and quality control. By connecting a micro-cuvette to the pipette tip, fluorescence detection is seamlessly integrated into the liquid handler workflow.
Sample and reagent dye volumes are reduced through the use of disposable cuvettes combined with an optimized workflow. Sample and reagent are dispensed and mixed in a 10 µL micro-cuvette. Fluorescent dye incorporation is monitored while mixing, and a reading of the stabilized signal is taken within seconds, minimizing overall workflow time.
The compact eviFluor Duo module, in SBS format, integrates into established liquid handling platforms and is compatible with disposable pipette tips of practically all manufacturers. Manufacturers can incorporate the eviFluor Duo into their applications as an OEM module.




 eviDense UV Photometer
eviDense UV Photometer
Product Highlights
- Absorbance and optical density measurements integrated seamlessly into liquid handler workflow
- Measurement at four wavelengths for nucleic acid concentration determination and purity check
- High accuracy and reliability thanks to consistent automated workflow and precise micro-cuvette technology
- Increased data integrity with automatic sample identification, data capture and transfer, ready for downstream processing
- Space-saving design with compact SBS footprint
- No loss of precious sample material for use in downstream processes
The Task
Automated genomics applications such as next-generation sequencing (NGS) strongly depend on the quality of the samples. Therefore, direct in-process integration of concentration and quality measurement is becoming an increasingly urgent need for users.
A module for the photometric analysis of samples, designed to be integrated into liquid handlers, is intended to meet this need.
The Challenge
On the one hand, the module should be small enough to fit within the space of a standard microtiter plate.
On the other hand, sample handling must be designed in such a way that the valuable sample is preserved and not discarded. The measurement should not only be accurate and cover multiple wavelengths, the measurement process should also be as cost-effective as possible. The measurement results must be of the same quality as those obtained with conventional standalone instruments and plate readers.
The Solution
The eviDense UV Photometer enables in-process DNA and RNA quantification and quality control. By connecting a micro-cuvette to the pipette tip, absorbance and optical density measurements are seamlessly integrated in the liquid handler workflow.
Disposable cuvettes, made of highly UV-transparent plastic, are at the heart of eviDense UV and enable intelligent sample handling without any loss of sample material. Connected to the pipette tip of a liquid handler, they are transported to the detector unit, where the sample is transferred into the cuvette and re-aspirated after measurement.
Using LED photodiodes, the detector unit measures at four wavelengths with minimal space requirements. This enables precise concentration determination while reliably identifying and quantifying impurities such as proteins or salts.
The eviDense UV module integrates into all established liquid handling platforms and is compatible with the disposable pipette tips of practically all manufacturers. Manufacturers can incorporate eviDense UV into their applications as an OEM module.




 WELLJET
WELLJET
Project Highlights
- Patented plate-handling mechanism
- Two instrument versions with shared key modules
- User friendly graphical user interface (GUI) via a touchscreen
The Task
We were approached to develop two next-generation instruments: a reagent dispenser and a dispenser stacker.
The main objectives were to achieve highly precise, reliable liquid dispensing for a wide range of microplate formats and to ensure compatibility with high-throughput laboratory workflows.
The Challenge
Development of the next-generation WELLJET Reagent Dispenser and Dispenser Stacker posed a series of technical challenges.
Due to the complexity of the new requirements, the project needed to start from scratch, as updating the previous generation of instruments (Viafill) would not have met the necessary performance standards.
The main challenge was to integrate a novel peristaltic pump technology, developed in parallel by the client, into two distinct instrument versions.
Additionally, it was critical to ensure flexibility in handling a wide range of microplate formats (from 6 to 1536 wells), while maintaining an ultracompact footprint suitable for use in laminar flow cabinets. Another challenge was balancing high-throughput demands with the need to minimize reagent waste, while delivering precise, non-contact dispensing for applications like ELISAs, genomic assays, and high-throughput screening.
The Solution
To address the ambitious project goals, we began the development of the WELLJET system from scratch, allowing for maximum flexibility in the design process. This approach was necessary to fully support the integration of the client’s innovative peristaltic pump technology, which was being developed in parallel. The primary focus for us was on system architecture and ensuring the compatibility of the core mechanical and software components. We made use of modular designs that could be reused across both the dispenser and stacker models.
Throughout the project, we worked closely with the client, not only delivering two fully functional prototypes but also providing a baseline firmware and software package. These prototypes included an intuitive GUI to streamline the user experience and ensure that the instruments were as user-friendly as they were technically advanced.
While the industrialization and design transfer were managed by the client, we continued to support these processes by sharing the extensive knowledge we had gained during the development phase. This ensured that the transition to mass production was smooth and efficient.
Our collaboration with the client resulted in the successful launch of both the WELLJET Reagent Dispenser and the Dispenser Stacker, which now set new standards for precision and reliability in liquid handling.
Find out more on the product website of Integra Biosciences AG.


 Workflow Demonstrator
Workflow Demonstrator
Project Highlights
- Creation of a functional and visually unique demonstrator within 4 months
- Innovative LED light concept to visualize workflow status - inspired by Molecular Cartography images
- Simulation of a realistic and interactive workflow using a wizard - integrated and available in the demonstrator
The Task
The objective of this project was to design and build a mockup for a revolutionary Molecular Cartography instrument. This simulates the workflow for gene expression analysis within the spatial and morphological context of a tissue sample – at single-cell resolution.
The mockup shows the workflow from a user interaction perspective, including key steps for sample preparation and measurement through a wizard, which is linked to specific drawer actions.
It provides a realistic user experience for demonstration at one of the most prestigious trade fairs in the industry.
The Challenge
One of the main challenges was creating a realistic yet out of the ordinary industrial design, the workflow demonstrator, a simple yet engaging user interface that matches the envisioned product.
Additionally, managing the tight timeline for design, assembly, and testing while ensuring collaboration between multiple teams, suppliers, and the client was a critical factor for the project's success.
The Solution
The solution is a visually appealing and workflow demonstrator that created the desired WOW effect at the trade show.
The luminous LED animations – inspired by molecular chartography – are the visual highlight of the product.
The mechanical design incorporates drawers for sample, consumable, and waste handling, while the user-friendly high-resolution, 19 inch through-glass touchscreen guides users through a simulated workflow.



 Fluoreye
Fluoreye
Project Highlights
- Fluoreye® is likely the world’s smallest DNA and RNA quantification device, is integrable, and is even retrofittable to existing Hamilton Robotics liquid handling robots.
- The product was invented by HSE’s innovation team and brought to a functional model within 6 months.
- Following the licensing contract, the final product was developed and transferred to manufacturing by HSE within 2 years by a small team of 5 people.
- The HSE invention gives Hamilton Robotics a competitive advantage while avoiding most of the technology risk.
The Task
To address a workflow break in the analysis of nucleic acid samples, we needed to invent and develop a fluorescence detector. It was crucial that the fluorescence detector could be picked up by a liquid handling probe and moved to the sample, instead of feeding the sample to the reader. Thereby, we had to achieve state-of-the-art detection performance while minimizing the footprint.
The Challenge
The challenges were to miniaturize the “flying” module and to achieve safe and wireless data communication. Only one SLAS/ANSI position on the robot’s deck could be used for the entire detection unit. The module also needed to integrate seamlessly into all Hamilton Robotics liquid handlers and their software suite and allow international distribution.
The Solution
The measurement unit is a very compact dual-channel confocal LED-driven optical device which is equipped with low-power Bluetooth® data communication and a chargeable battery pack. It is encrypted and paired with NFC technology and an optical reference is integrated for functional checks.
Small sample volumes in a microwell plate can be quantified to ensure processes like library prep for NGS become predictable and reliable. All applicable international standards for wireless communication, including EMC, were tested and met. The software functionality covers all use cases, namely QC after manufacturing, installation, use, and maintenance.
Throughout the entire development time Hamilton Robotics, the licensor, was involved and kept up-to-date and they ultimately took over the manufacturing.
Find out more on the product's website.



 Ampha P20
Ampha P20
Project Highlights
- The Ampha P20 is the world’s first fully portable pollen analyzer.
- The modular design approach allows modules to be reused in other Amphasys platforms.
- We went from concept to series product in about 2 years.
- We had a small but highly efficient team of around 5 HSE employees.
The Task
We needed to develop a small, portable, and easy-to-use pollen analyzer based on impedance flow cytometry technology. Importantly, the device should enable rapid measurements through a simplified user interface and automated data analysis. The intended portability and ease of use will drastically increase the efficiency of pollen analysis in plant breeding, production research, and seed production.
The Challenge
The big challenge was to pack the complex flow cytometry technology into a small, robust, and battery-powered portable device while ensuring maximum user and service friendliness. Combining the extremely sensitive measurement system with a solid-plastic case resulted in additional design challenges, for example in terms of mechanical tolerances and EMC compliancy. Furthermore, the accuracy of the measurement could only be guaranteed if the dead volumes in the fluidic path are very small.
The Solution
We integrated the liquid-handling system into a manifold. The component, which is milled in synthetic resin, contains the entire channel system through which the samples and reagents flow on an area of just a few square centimeters. This minimizes the dead volumes. In addition to the manifold, the device contains only three tubes, one pressure sensor and four valves for the fluidics.
The EMC compliancy of the entire device was achieved through intensive testing and iterative design improvements.
Sophisticated power management allows use of a lithium battery pack that can be recharged directly in the device or exchanged if needed (cordless screwdriver principle). A fully charged battery enables about 6 hours of full operation in the field.
Find out more on the product's website.

 Ampha Z40
Ampha Z40
Project Highlights
- The Ampha Z40 is a more compact and powerful enhancement of the proven Ampha Z32.
- Thanks to a modular design approach, we were able to reuse modules from other Amphasys platforms, especially the well-thought-out manifold of the mobile Ampha P20.
- An eye-catching design increases the brand awareness of Amphasys.
- We went from concept to series product in about 1 year.
- We had a small but highly efficient team of around 5 HSE employees.
The Task
Our objective was to develop a very compact state-of-the-art benchtop instrument for pollen analysis and research applications, based on impedance flow cytometry technology. The Ampha Z40 is supposed to be a more enhanced version of the proven Ampha Z32 benchtop device and shall enable more performance-intensive applications.
The Challenge
To achieve a more compact device, our first priority was to miniaturize the fluidics. A more powerful chip and additional interfaces were required to enable performance-intensive research applications. In addition, the device needed to be operated either via the embedded PC with an external monitor or via a separate PC.
The Solution
The fluidic system, including the core module with an ultra-compact manifold, was an adaptation of the corresponding module of the mobile Ampha P20. The Ampha Z40 has a unique industrial design allowing maximum usability and is an eye-catcher in any lab. The enhanced instrument automates all steps from measurement to evaluation of the pollen. It is the ideal instrument for collecting information on pollen lines and genetic material from early research to commercial seed production. It supports process optimization with cost reduction and improvements for higher seed yield.
Find out more on the product's website.
 Ampha X30
Ampha X30
Project Hightlights
- The Ampha X30 represents the next generation of cell analyzers for bioprocessing applications, such as bacteria, yeast, or somatic cells.
- Thanks to a modular design approach, we were able to reuse modules from other Amphasys platforms to some extent and develop them further.
- An eye-catching design increases the brand awareness of Amphasys.
- We went from concept to series product in about 1 year.
- We had a small but highly efficient team of around 5 HSE employees.
The Task
The Challenge
The challenge was to find a way to adapt the proven Ampha Z40 technology for bacteria analysis. Compared to the pollen application, the bacteria application has different requirements including smaller fluidic channels to enable accurate impedance measurements. The instrument also requires a more stringent grounding concept and EMC protection measures to ensure reliable measurements.
The Solution
We were able to reuse the manifolds of the P20 and Z40 as a basis for adaptation to the special requirements of bacteria, yeast, and various somatic cells. We made major modifications to the manifold, such as adding new channels and implementing new valves. Like the Z40, the Ampha X30 has a unique, eye-catching industrial design allowing maximum usability and offering a high brand recognition value. The instrument automates all steps from measurement to evaluation of the bacteria. It allows simultaneous measurement of cell count and cell viability and provides crucial information about the status of the cells, like metabolic activity. No biomarkers or dyes are needed for sample preparation which reduces the costs of the analysis significantly.
Find out more on the product's website.

 QIAcube Connect
QIAcube Connect
Project Highlights
- Reduction of production costs
- Improved usability
- Additional functionality
The Task
Our task was to enhance user-friendliness and efficiency of the QIAcube Connect in molecular biology labs. The instrument update aimed to introduce advanced digital features and connectivity, including tablet integration for real-time monitoring and error handling.
Additionally, our goal was to reduce production costs for the QIAcube Connect compared to the original QIAcube. We also aimed to maintain automation of existing protocols. The user-friendly software and touchscreen interface should be designed to guide users through setup, minimizing the risk of errors. Furthermore, we were asked to ensure compliance with the latest safety regulations.
The Challenge
We faced the following challenges during the development of QIAcube Connect. These included ensuring global connectivity while adhering to the strict security standards of different countries and ensuring data security. One challenge was to reduce production costs while adding functionality such as a UV LED and a larger screen and maintaining high-quality performance of the instrument.
In addition, developing user-friendly software and a touchscreen interface that meet safety and compliance standards was crucial. Similarly, strict adherence to the latest safety regulations was a prerequisite that required thorough testing and validation.
The Solution
Our successfully overcame the challenges of developing QIAcube Connect. To ensure global connectivity and comply with safety standards, country-specific connectivity packages have been developed that eliminate the need for individual certifications.
Advanced manufacturing techniques, production relocations and process optimizations were used to optimize costs.
User-friendly software has been developed that meets safety and compliance standards and is even IVD-compliant in the CIAcube Connect MDx version. Safety improvements were made, particularly to the electronics. Our innovative solutions ensured the success of QIAcube Connect by effectively overcoming every challenge. These included cost-effective production, software expertise, safety improvements and global compliance, resulting in an improved device for molecular biology laboratories.
QIAGEN QIAcube Connect product website
QIAcube Connect MDx
Project Highlights
- IVD and research mode
- Traceability and process control
- User management
The Task
Transforming the QIAcube Connect into the QIAcube Connect MDx primarily focused on developing specialized software for molecular diagnostics.
This process was guided by compliance with essential regulatory standards. These included ISO 13485 for medical device manufacturing, the IVDR in Europe for IVDs, FDA 21 CFR Part 820 in the USA, CE marking for European market compliance, and CLIA standards in the USA for laboratory testing. Adhering to these standards was critical to ensure the device's safety and efficacy in clinical use.
The Challenge
The challenge was to integrate IVD software into QIAcube Connect MDx, offering protocol choice via a user-friendly touchscreen. Detailed run reports include kit information, sample status, and maintenance information, aiding sample tracking and quality control.
User management enhances security with role-based access. This streamlined system meets the needs of IVD applications effectively.
The Solution
The implemented solution involves integrating new IVD) software into the QIAcube Connect MDx. It allows users to select between IVD (in vitro diagnostics) and MBA (molecular biology application) protocols prior to starting procedures.
It offers a user-friendly touchscreen interface for step-by-step guidance.
Detailed run reports include critical data such as kit expiration date, sample status, location, and instrument maintenance information. This facilitates sample tracking and quality-control compliance with IVD requirements.
The software also enhances data protection and user tracking through user management capabilities, with administrators assigning controlled access roles and optimizing efficiency and security in IVD applications.
QIAGEN QIAcube Connect MDx product website
 TubeSealer Module
TubeSealer Module
TubeSealer module enables automated qPCR tube sealing
The TubeSealer is a module that performs fully automated sealing of qPCR reaction vessels. It consists of a proprietary bead dispenser (1,2) that dispenses beads into each selected vessel using a plunger. The plunger applies a defined force to the beads to achieve maximal tightness.
Pure reliability
The bead dispenser holds up to 360 beads of 4 mm diameter, which are dispensed at a 100% dispensation rate securing high reliability. A plunger presses the beads into each vessel, efficiently sealing them for the subsequent qPCR. The design of the instrument inherently prevents any cross-contamination between the samples. To further increase the safety of the process, the tightness of the seal is checked and confirmed by a built-in optical sensor. To prevent any degradation of the samples, a cooling plate can be positioned below the sample plate. The only manual intervention required for the process is supplying the samples and selecting a suitable program. As an option, a load-check of the vessels can be performed if necessary. All these features combined, ensure that the integrity of the samples is maintained throughout the PCR process.
Exceptional flexibility
The TubeSealer is incredibly flexible when it comes to throughput, vessel type, vessel dimensions, liquid volume and communication interface. The instrument can be used equally well for any throughput; low, medium or high. It is compatible with a wide range of tube sizes and designs, such as single tubes, tube strips, 96-well plates, discs, conveyor belts, etc. The bead dispenser can be adapted to different bead sizes required for various diameters of the tubes. The instrument is also compatible with different interfaces, so that data can be transferred via cloud, WLAN and bluetooth. The instrument is designed in the way that is possible to seamlessly integrate instrument software compliant with the SiLA standard for software control. These properties truly demonstrate the flexibility of the automated sealing process carried out by the TubeSealer module.



 QIAcube HT
QIAcube HT
Mid- to high-throughput nucleic acid purification
QIAcube HT performs mid- to high-throughput automated purification of nucleic acids in a 96-well format. The instrument is based on silica membrane technology and thus yields high quality nucleic acids intended to be used for molecular biology applications.
Safe and standardized
The QIAcube HT can be used for purification of a wide range of compounds such as DNA, RNA and miRNA from virtually all sample types, including cells, tissues, stool samples, raw or processed food material as well as bacteria and viruses. In order to keep a safe environment within the instrument, a HEPA filter is used to purify the air and maintain a positive air pressure while the UV light decontaminates the worktable and prevents cross-contamination. There is also a tip ejection feature, making sure that used tips are collected externally thereby keeping the internal workspace clean, as well as dedicated plasticware ensuring reliability and convenience.
Simple integration in workflows
The straight-forward purification procedure can seamlessly be integrated in a number other workflows. The eluted nucleic acids samples are collected in tubes that are compatible with downstream applications carried out in QIAgility and different detection systems. Purification steps can easily be included into more complex workflows. The simplicity of the procedure is also enhanced by its intuitive software, where the graphical user interface simulates the instrument worktable and allows a run to be initiated with only a few mouse clicks. The data from each run as well as bar codes and other specific information is easy to manage and can readily be integrated in the Laboratory Information Managements Systems (LIMS). The combination of being a high quality, high-throughput bench-top instrument and having a noticeably compact design, makes it a valuable option for biosafety level laboratories.

 QIAsymphony SP
QIAsymphony SP
Case Study QIAsymphony
The Challenge
Decentralized molecular testing by enabling medium throughput laboratories. Molecular testing is largely centralized in specialized laboratories, reference labs and large hospitals. Sample types are very diverse, come in very different containers and need to be tested for a broad range of diseases.
The Approach
In order to accommodate the various setups and demands of the laboratories a modular system design with sample preparation, assay setup and analysis was chosen. Ease of use and quick setup of the system was achieved with a disposable, pre-filled reagent cartridge design loaded into an easily accessible drawer. The sample input and output area can be loaded with individual tubes or microplates. The system is operated via touchscreen. The core sample processing module can be extended with either an assay setup module or an automated PCR cycler unit for up to 140 analyses per run.
The Goal
Build a system that accommodates for the immense variety of setups of medium throughput testing labs. All samples types and input formats will be accepted by the system. 1 to 96 samples, input volumes from 100 ul to 1 ml and processing of any type of PCR assay gives the operator highest flexibility. Despite its flexibility, the system will be set up in less than 15 min. and run from start to finish without user intervention.
The Result
2000 units have been sold globally so far. QIAsymphony is the undisputed standard in automated nucleic acid sample processing for the medium throughput laboratory.
Case Study QIAsymphony Cartridge
The Challenge
Biological samples are extremely divers requiring a large number of methods and chemistries to purify nucleic acids and provide them in a format for further processing.
The Approach
Individual troughs are deep-drawn and heat sealed for low cost and long-term storage. The heat seal withstands aggressive chemicals for an indefinite storage period. Troughs are held together with a frame for easy handling. A 2D barcode will identify the individual troughs. A plastic disposable piercing device is used to punch holes to allow the pipetting head to pick up reagents. Low volume enzymes are stored in a separate rack for cold storage and can be attached to the through rack to build one handling unit.
The Goal
Build a low cost disposable, pre-filled, IVD compliant cartridge that accommodates dozens of different magnetic bead chemistries. The cartridge is filled, assembled and sealed in an automated production. Will be opened automatically by the QIAsymphony system and can be resealed if not completely used. The shelf life of the cartridge is > 1 year. Productions cost are equal to or lower than comparable spin column products.
The Result
The QIAsymphony cartridge is an extremely cost-effective and reliable reagent storage container used thousand-fold in diagnostic laboratories around the world.
 EZ1 Advanced XL
EZ1 Advanced XL
Automated nucleic acid purification
The EZ1 Advanced XL is a desktop instrument designed to perform automated isolation and purification of nucleic acids, such as eukaryotic DNA, mRNA, total RNA as well as viral RNA and DNA. The system is based on proven magnetic bead technology and can process up to 14 samples in each run in 20 minutes. The process consists of reading the reagent and sample information with a bar code scanner, lysing the samples, binding the nucleic acids to the beads, washing and finally eluting the nucleic acids.
Safety and accuracy
The integrity of the samples and the reproducibility of the procedures are ensured by the design of the instrument. A UV light is used for decontamination, efficiently eliminating any Gram-positive and Gram-negative bacteria inside the instrument. In addition, a sensor makes sure the instrument door is closed during sample preparation, thereby further protecting sample integrity. Manual intervention is kept to a minimum by providing EZ 1 Kits containing all necessary reagents in a format that can be processed automatically by the instrument, from the opening of reagent cartridges to the elution of nucleic acids. Furthermore, the instrument contains high-precision syringe pumps with tip adapters holding filter tips, which can aspirate and dispense 50–1000 µl liquid with exceptionally high accuracy.
Traceability and versatililty
Samples and consumables can easily be tracked using the bar code reader and the manual keyboard. All system parameters and run data are stored in the system and are readily accessible. The instrument will generate a pdf and a csv result file after each run. The report files can either be exported to a LIMS (Laboratory Information Management System) or other programs for any downstream applications. The provided EZ1 Kits are available for a large number of applications used in forensics, biomedical research, molecular diagnostics and gene expression analysis.


 QIAgility
QIAgility
High-precision automated PCR setup
The QIAgility is a compact benchtop workstation used to automatically set up PCR assays with exceptionally high-precision. Since all manual pipetting steps are eliminated, QIAgility can ensure both reproducibility and increased productivity. It features a single channel pipetting head, delivering fast and safe transfer of liquids, and features dynamic liquid-level sensing with a minimum detection volume of 10 µl.
Pure pipetting accuracy
The reproducible and highly accurate PCR setup mainly relies on the exceptionally high quality of the dispensations, which range between 1 µl and 200 µl. This is enabled by the QIAgility Setup Manager software and the precision of the dispensed volumes has been verified by repeated tests in various settings. The high sensitivity of the system is ensured by its ability to sense the meniscus during pipetting and retracting the tip during dispensation, thereby increasing the accuracy and minimizing any carry-over. If the liquids are viscous, the pipetting speed will be adapted accordingly. Tests with serial dilutions have proven that the linearity of system is exceptional, with a regression coefficient of >0.999. Moreover, the software provides step-by-step guidance and automatically calculates all mixes, so that no pipetting steps need to be programmed. Any contamination of the samples is prevented by an optional UV light and the HEPA filter system ensures positive pressure of clean air within the instrument. In addition, all used tips are ejected externally, so that no waste is accumulated on the worktable.
Flexibility and applications
The PCR assay setup performed by QIAgility can easily be integrated into various workflows. Almost all tube and plate formats are supported, such as 96-well and 384-well plates, but also Rotor-Discs for the Rotor-Gene® Q real-time PCR instrument. For added convenience, a laptop computer with intuitive software is provided. The user can program various pipetting applications, such as normalization of DNA and RNA concentration, transfer of liquid samples between different tube formats, serial dilutions with variable dilution ratios, restriction digest setups, sample pooling and selective pipetting from archived sample banks.


 QIAsymphony AS
QIAsymphony AS
Case Study QIAsymphony
The Challenge
Decentralized molecular testing by enabling medium throughput laboratories. Molecular testing is largely centralized in specialized laboratories, reference labs and large hospitals. Sample types are very divers, come in very different containers and need to be tested for a broad range of diseases.
The Goal
Build a system that accommodates for the immense variety of setups of medium throughput testing labs. All samples types and input formats will be accepted by the system. 1 to 96 samples, input volumes from 100 ul to 1 ml and processing of any type of PCR assay gives the operator highest flexibility. Despite its flexibility the system will be setup in less than 15 min. and run from start to finish without user intervention.
The Approach
In order to accommodate for the various setups and demands of the laboratories a modular system design with sample preparation, assay setup and analysis was chosen. Ease of use and quick setup of the system was achieved with a disposable, pre-filled reagent cartridge design loaded into an easily accessible drawer. The sample input and output area can be loaded with individual tubes or micro- plates. The system is operated via touchscreen. The core sample processing module can be extended with either an assay setup module or an automated PCR cycler unit for up to 140 analyses per run.
The Result
2000 units have been sold globally so far. QIAsymphony is the undisputed standard in automated nucleic acid sample processing for the medium throughput laboratory.
Case Study QIAsymphony Cartridge
The Challenge
Biological samples are extremely divers requiring a large number of methods and chemistries to purify nucleic acids and provide them in a format for further processing.
The Goal
Build a low cost disposable, pre-filled, IVD compliant cartridge that accommodates for dozens of different magnetic bead chemistries. The cartridge is filled, assembled and sealed in an automated production. Will be opened automatically by the QIAsymphony system and can be resealed if not completely used. Shelf life of the cartridge is > 1 year. Productions cost are equal or lower than comparable spin column products.
The Approach
Individual troughs are deep-drawn and heat sealed for low cost and long term storage. The heat seal withstands aggressive chemicals for an indefinite storage period. Troughs are hold together with a frame for easy handling. A 2D barcode will identify the individual troughs. A plastic disposable piercing devise is used to punch holes to allow pipetting head to pick up reagents. Low volume enzymes are stored in a separate rack for cold storage and can be attached to the through rack to build one handling unit.
The Result
The QIAsymphony cartridge is an extremely cost-effective and reliable reagent storage container used thousand-fold in diagnostic loaboratories around the world.
 Rotor-Gene Q
Rotor-Gene Q
Precise and robust real-time PCR
The real-time PCR-cycler Rotor-Gene Q is designed to be used for real-time and end-point thermal cycling using polymerase chain reaction (PCR) and high-resolution melting analysis (HMR) for various molecular biology applications. It has a unique design with a rotor that spins the samples, which minimizes the optical and thermal variation. The instrument’s stability in combination with its fast ramping rates and short equilibrium times renders Rotor-Gene Q highly suitable for fast, sensitive and accurate quantitative analyses.
User-friendly design and software
With its small, light and robust design there is hardly any need for maintenance. There is a lifetime guarantee for the LEDs and no need for optical calibration or cleaning of sample blocks. Also, the rotary design prevents any condensation or bubbles forming in the reactions. Rotor-Gene Q can be used with either tubes or Rotor-Discs for faster handling of high-throughput workflows. In addition, it features intuitive software, such as the Q-Rex Software, which supports all analysis procedures from basic to advanced algorithms, and its extension software Rotor-Gene ScreenClust HRM Software, which enables HRM analysis for genotyping, mutation screening, pathogen typing and quantitative methylation analysis. Furthermore, the software REST software 2009 takes the different PCR efficiencies of the gene of interest and reference genes into account and provides analysis of e.g. up- and downregulation for gene expression studies.
Widest optical range — even more applications
Rotor-Gene Q has an unrivaled optical range with up to 6 channels spanning from UV to infrared wavelengths and can therefore be used with a number of different fluorophores. The software also allows addition of new wavelength combinations so that it will be compatible with new dyes to be used in future applications. Today, the instrument can be used for applications such as gene expression analysis, genotyping, pathogen detection, mutation analysis, DNA methylation analysis and miRNA research.


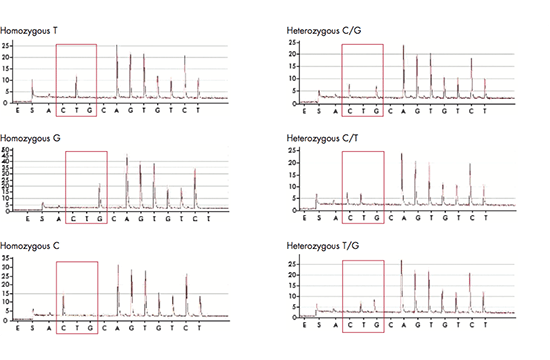
 PyroMark Platforms
PyroMark Platforms
Quantifying sequence variations with pyrosequencing
The PyroMark platforms are used for rapid and accurate quantification of genetic and epigenetic DNA modifications using pyrosequencing assays. They can be used for advanced methylation, mutation and SNP quantification but also for verification and validation of samples from NGS and array experiments. The PyroMark platform consists of PyroMark Q24, PyroMark Q24 Advanced, PyroMark Q48 Autoprep, and PyroMark Q96 ID. PyroMark Q24 can analyze up to 24 samples in 15 minutes, can read up to 80 bp and performs mutation analysis and resistance typing. PyroMark Q24 Advanced shares those features but can read up to 140 bp and perform more complex mutation analyses, as well as epigenetic (CpG and CpN) analyses and microbial typing. PyroMark Q48 Autoprep with automated sample preparation is used for similar applications and can read equally long sequences, but can analyze up to 48 samples at a time. Finally, PyroMark Q96 ID has the capacity of analyzing up to 96 samples in each run.
From pyrosequencing to analysis reports
The pyrosequencing reaction relies on the release of light signals, where the amount of light is directly proportional to the number of incorporated nucleotides. The four nucleotides A, C, G and T are dispensed in a predefined order and as the DNA is elongated, pyrophosphate is released and converted to ATP, which is used by luciferase to generate light. The result of each dispensation of nucleotides is shown in the pyrogram, so if for example 3 nucleotides are incorporated (e.g. AAA), three ATPs are generated giving rise to triple peak in the pyrogram. If no nucleotides are incorporated, there is no light signal and thus no peak. The PyroMark software can be set up in different analysis modes in order to target analysis of for example SNPs, CpGs or perform base-calling of unknown sequences. When the analysis has been finalized, the software will generate comprehensive reports displaying the results as pyrograms as well as presenting statistics such as methylation percentage, allele frequencies and other analysis results.



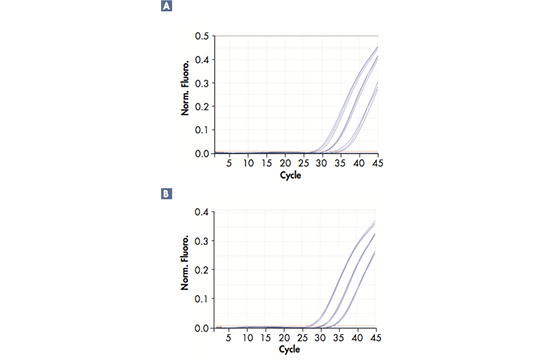
 GeneReader NGS
GeneReader NGS
Case Study GeneReader
The Challenge
The capacity to sequence all 3.2 billion bases of the human genome has increased from 1.3 human genomes sequenced annually to 18,000 human genomes a year within a decade. Despite this improvement sequencing a genome is still an extremely complex procedure, which prevents its broad use in routine human diagnostics.
The Goal
Build a complete NGS workflow with seamlessly integrated automated components offering ease of use and efficiency from sample to result. Provide actionable insights with validated gene panels and fully integrated bioinformatics.
The Approach
The GeneReader workflow includes the following 6 processes: sequencing primer hybridization, flow cell preparation, reagents preparation, experiment set-up, flow cell loading and run start and post-run maintenance wash.
The Result
The GeneReader is the only integrated NGS workflow from sample to insight.
 QIAxcel Connect
QIAxcel Connect
Project Highlights
- Second optical channel for higher sensitivity
- Innovative filter changer for dye emission
- Connectivity package for real-time status monitoring
The Task
As the complexity and demands of downstream applications grow, it is vital that DNA and RNA quality control measurements deliver sensitive, high-resolution results. Our task was to significantly increase the sensitivity compared to the previous product ‘QIAxcel Advanced’. We have achieved this by adding a second optical channel in the new generation model ‘QIAxcel Connect’.
In addition, the instrument update also included a new housing and advanced connectivity for real-time status monitoring.
The Challenge
One of the main challenges we faced was the lack of space inside the instrument. The filter and filter changer for the second optical channel needed to be small enough to fit into the limited space to avoid moving any of the other internal components in this complex system. We also had to ensure global connectivity while adhering to the strict security standards of different countries and ensuring data security.
The Solution
Traditionally, filters are usually changed using a filter wheel, but this was not a viable solution due to the lack of space inside the instrument. We successfully overcame this challenge by developing an innovative filter slider, which requires much less space. The filter slider has a filter at each end and the QIAxcel Connect selects the correct filter by reading the information on the gel cartridge smart key.

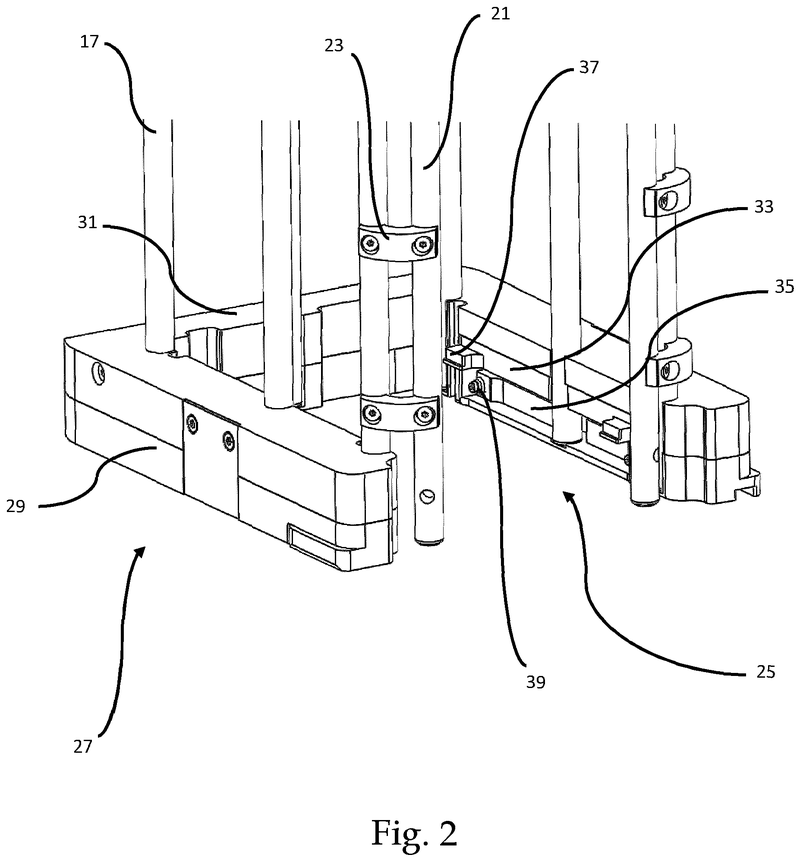
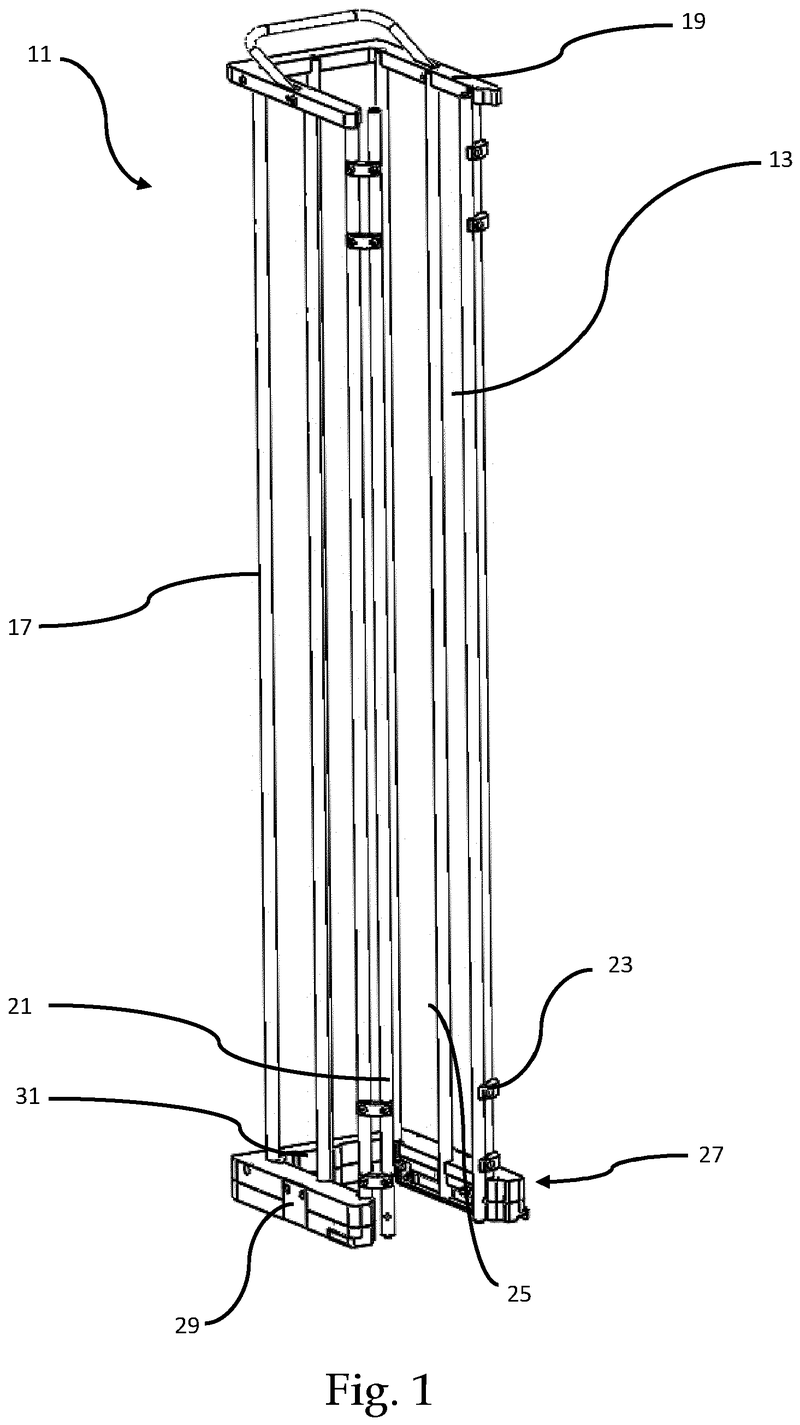
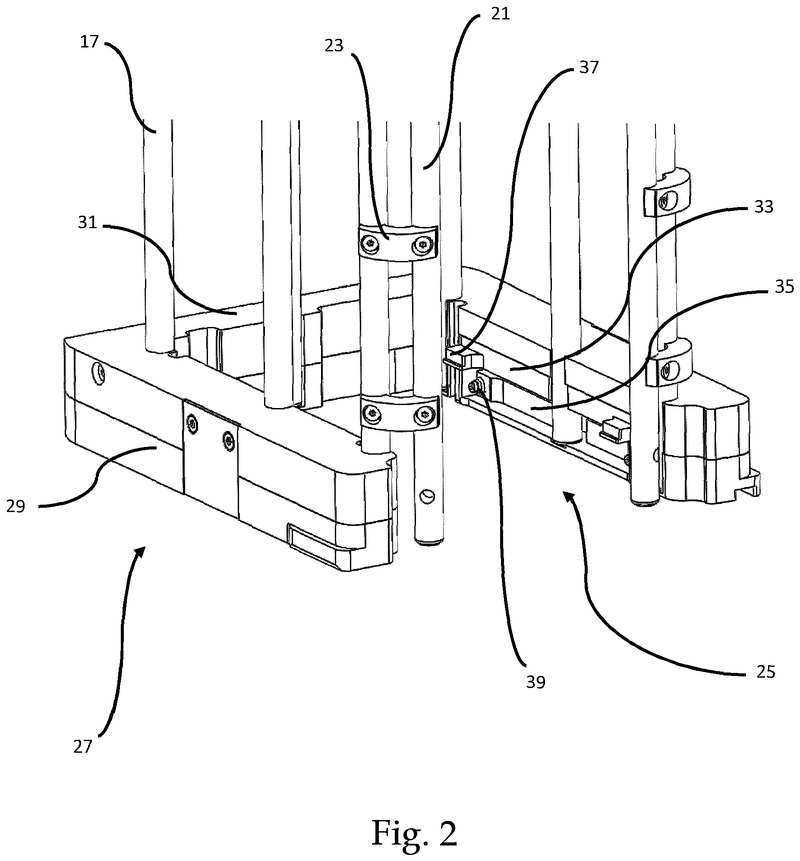
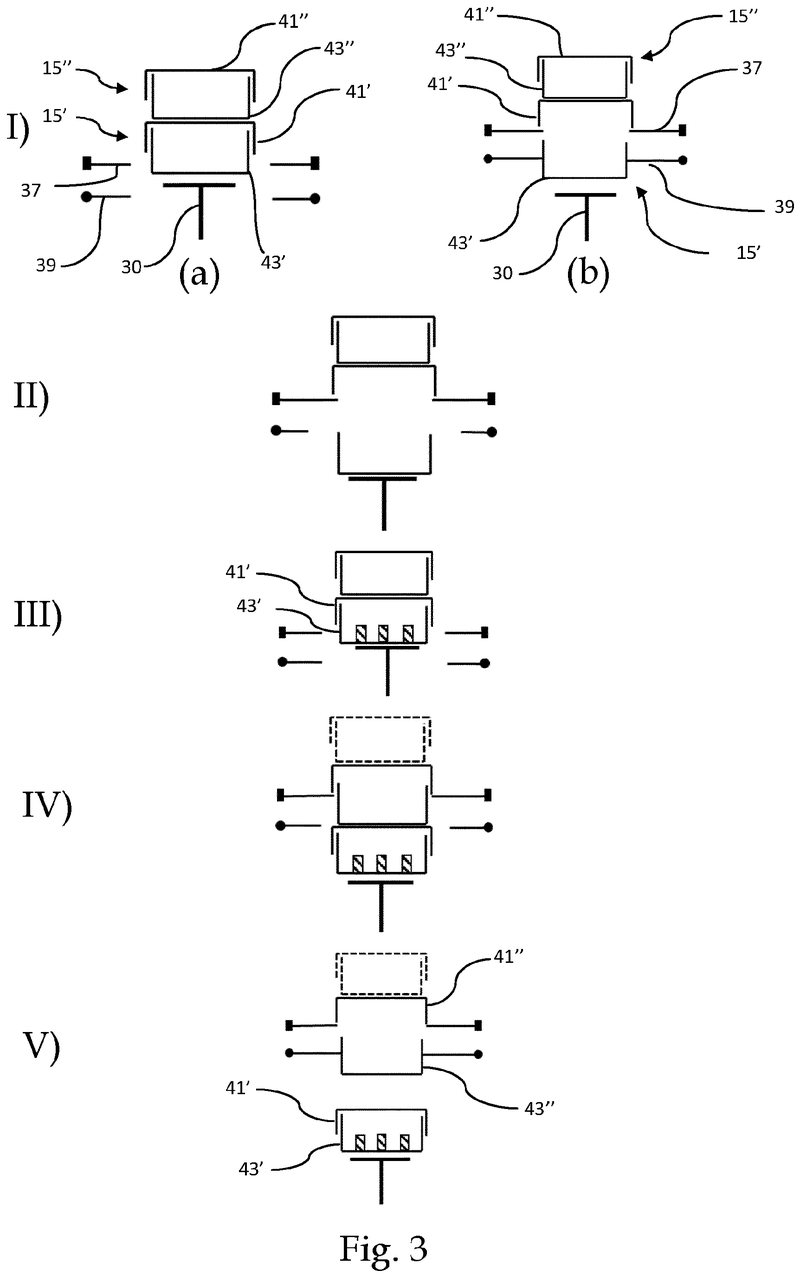
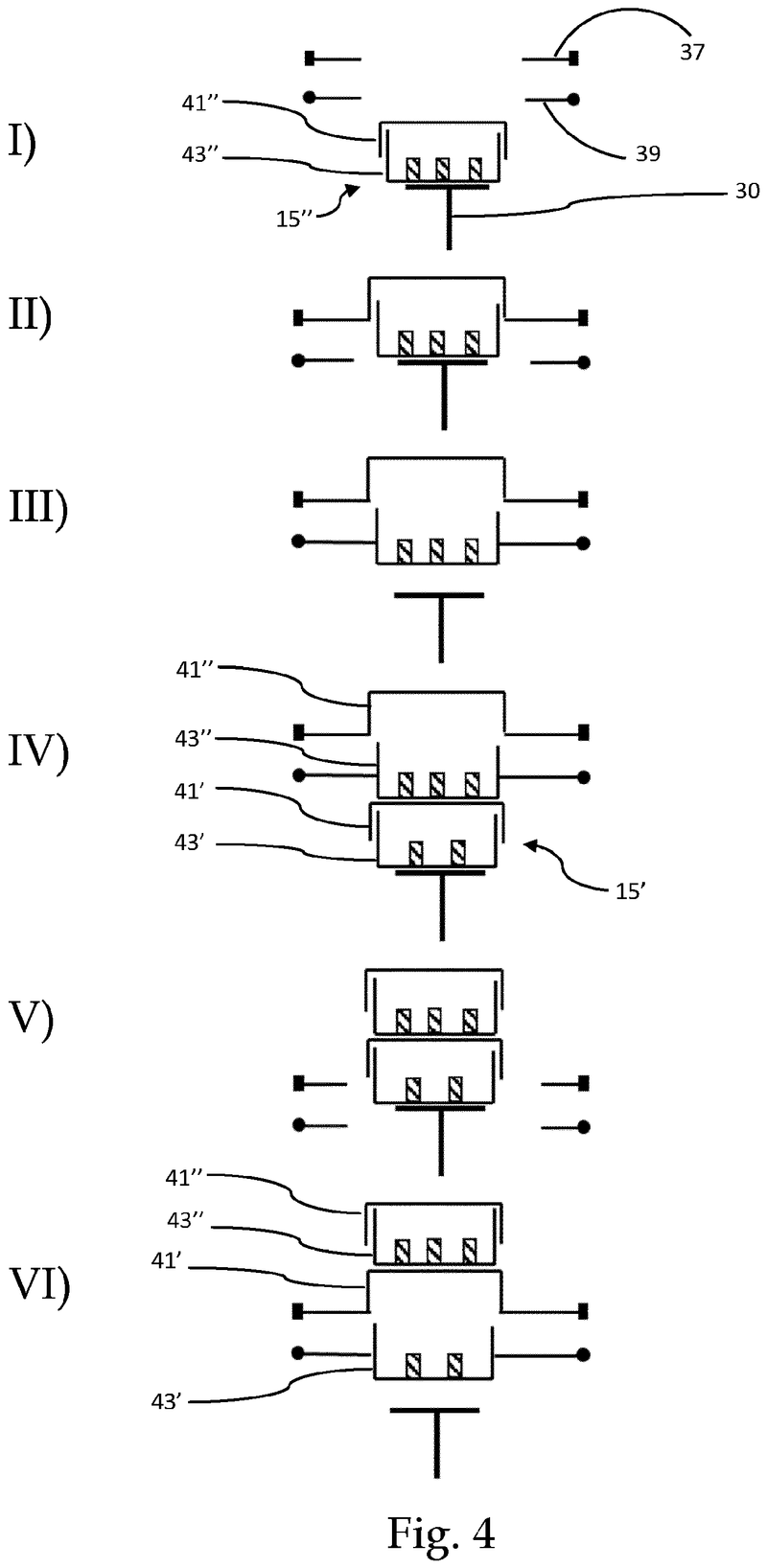
Plate handling
Stacking device for microtiter plates
Highlights
- Innovative plate stacking mechanism
- Reliable separation of microplates and lids
- Compatible with virtually all kinds of microplates
The Challenge
The challenge was to enable reliable separation and transfer of individual microplates stacked in a tower to an instrument for automated filling followed by restacking in the output tower.
This is especially difficult when working with microplates with lids as the lid must be removed and then replaced after filling. As lids vary greatly in their interface with microplates, this was an additional challenge we had to overcome.
The Solution
The problem was solved very elegantly by combining the separation mechanism and the stacking tower.
By fixing the microplate or lid using grippers with a form-fitting connection, the weight of the entire stack can be stably supported on the upper axis of the separation mechanism.
Because the upper separation mechanism supports the weight of the stack of microplates resting on it, the lower separation mechanism axis only needs to fix one microplate in place via a frictional connection.
Our separation mechanism works perfectly with all the plates we have tested, taking into account the wide variety of microplates and lids that exist. Separation and transfer of microplates for automated filling is reliable, effective, and fast.